Reconstructing a Sanskrit text
Charles Li
Last updated: 6 October 2023.
This document is a companion to the sanskrit-alignment GitHub repository, where you will find open source software developed to work with multi-witness Sanskrit texts. The software has been used to reconstruct Bhartṛhari's 5th-century Dravyasamuddeśa along with its 10th-century commentary, which serves here as the example text for the tutorial.
The tutorial assumes some knowledge of both Sanskrit philology and phylogeny. For a short, gentle introduction to phylogeny, see Baldauf 2003,
Phylogeny for the faint of heart: a tutorial.The matrix-editor had its beginnings in 2019, as part of the project,
A Cultural and Textual History of Sanskrit Riddle-Tales: A Digital Humanities Approachat the University of British Columbia, led by Adheesh Sathaye, supported by the Social Sciences and Humanities Research Council. helayo, the sequence alignment package, was initially made possible by the Government of Canada, through the COVID-19 Canada Emergency Response Benefit in 2020. Finally, thanks to rsync.net for providing exceptional cloud backup services.
This document is licensed under a Creative Commons Attribution 4.0 International License. Its change history can be accessed through GitHub as part of the sanskrit-alignment GitHub repository. It is under active revision. If you have any comments, suggestions, corrections, or other changes, don't hesitate to contact me or to create an issue or submit a pull request.
The software has been published in the Journal of Open Source Software:
Li, Charles. 2022.
helayo: Reconstructing Sanskrit texts from manuscript witnesses. Journal of Open Source Software 7(71), 4022. https://doi.org/10.21105/joss.04022.
Table of Contents
tl;dr
- Transcription
- transcribe your manuscripts in TEI XML. Each file needs to have a siglum (
<idno type="siglum">), and each paragraph or verse to be collated needs to have anxml:id. See saktumiva.org for more details. - export your transcriptions using the converter. Alternatively, if you are on saktumiva.org, use the
Export to FASTToption. For this tutorial, sample TEI XML transcriptions from the Dravyasamuddeśa can be used, and FASTT files can be found in theexample/fastt/directory.
- transcribe your manuscripts in TEI XML. Each file needs to have a siglum (
- Alignment
- download the appropriate
helayobinary from thehelayo/dist/directory, or build it from source using the Haskell Platform. - run
./helayo -x substitution_matrix.csv [input.fas] > [output.xml]for each.fasfile. - open your
.xmlfiles in the matrix-editor and fix any big alignment errors. You can also useEdit -> Group all words. Save each file individually. - open all your
.xmlfiles in the matrix-editor at once (use Ctrl-Click or Shift-Click to select multiple files). Export to NEXUS.
- download the appropriate
- Building a tree
- open your NEXUS file in tree-building software (like SplitsTree5).
- build a tree and root it.
- export the tree as NeXML.
- Reconstructing a text
- open the NeXML tree in the matrix-editor.
- make sure that the texts are normalized (
View -> Normalized). - click on the node that you want to reconstruct (probably the root).
Introduction
Karl Lachmann's stemmatic method has often been derided as overly mechanical
Trovato 2014, 79, 82. by its opponents. But, in view of our ability to automate mechanical tasks using computers, this is, decidedly, a great virtue. That is not to say that a well-trained philologist is no longer needed; as this document will show, every step of the process requires careful decision-making on the part of the scholar. However, by offloading much of the work onto computer software, each automated task becomes reproducible. As you will see in this tutorial, given some data (i.e., a set of transcriptions), any scholar will be able to reproduce the resultant text, and to easily critique the philological decisions that led up to it.
There are four main steps in this tutorial:
- Transcription, in which each witness is carefully and diplomatically transcribed,
- Alignment, or Collation, in which the similarities and differences between each witness are tabulated,
- Building a tree, in which the alignment is used to infer a process of evolution, and
- Reconstructing a text, in which a critical text is created, based on the tree.
In this tutorial, we will be using my edition of Bhartṛhari's Dravyasamuddeśa with Helārāja's 10th century Prakīrṇaprakāśa commentary as an example. All manuscript transcriptions are available at https://saktumiva.org/wiki/dravyasamuddesa/.
The GitHub repository
You will need to download the files in the GitHub repository. The easiest way to do this is to download a zip file; click the green Code
button. Then extract the zip file into your working directory. These are the relevant subdirectories:
docs/— the location of this tutorialexample/— containing the example files used in this tutorialexample/fastt/— FASTT files exported from saktumiva.orgexample/xml/— those files aligned, which can be opened in thematrix-editorexample/trees/— phylogenetic trees generated from those alignments, which can also be opened in thematrix-editor
helayo/— the alignment tool (see Alignment)matrix-editor/— an interface to view/edit alignments, export them to be used with phylogenetic tree-building software, and reconstruct texts based on those trees
Transcription
 Rajasthan Oriental Research Institute, MS 4781.
The most arduous task in this process is the collection of witnesses and their transcription. Previously, transcribing each witness individually was considered to be too daunting a task; when a text is collated by hand, the editor only notes the differences between a witness and their edition. But in this process, a lot of information is lost; orthography and punctuation are ignored, and any variant readings that are considered insignificant are not mentioned. But with computer-aided collation, we can create diplomatic transcriptions of our witnesses and then automatically filter out variations that we wish to ignore. In this tutorial, all of the manuscripts have been transcribed in IAST using TEI XML. They have been uploaded to saktumiva.org, where you can automatically generate a critical apparatus comparing one version of the text to the others. For more information on how this works, see Li 2017 and Li 2018.
Rajasthan Oriental Research Institute, MS 4781.
The most arduous task in this process is the collection of witnesses and their transcription. Previously, transcribing each witness individually was considered to be too daunting a task; when a text is collated by hand, the editor only notes the differences between a witness and their edition. But in this process, a lot of information is lost; orthography and punctuation are ignored, and any variant readings that are considered insignificant are not mentioned. But with computer-aided collation, we can create diplomatic transcriptions of our witnesses and then automatically filter out variations that we wish to ignore. In this tutorial, all of the manuscripts have been transcribed in IAST using TEI XML. They have been uploaded to saktumiva.org, where you can automatically generate a critical apparatus comparing one version of the text to the others. For more information on how this works, see Li 2017 and Li 2018.
When producing your transcriptions, each witness should have a siglum (<idno type="siglum">). Then, each paragraph or verse (<p> or <lg>) should have a unique xml:id, so that the alignment software knows how to collate them. For examples, see the TEI XML transcriptions of the Dravyasamuddeśa.
Alignment
Saktumiva.org generates a critical apparatus using a pairwise sequence alignment algorithm. This is quick, and suits its purpose; in a critical apparatus, we are mainly interested in differences between our base text and each of the other witnesses. However, in order to create a phylogenetic tree, we want to align our texts using a multiple sequence alignment algorithm, which is more accurate but much slower.
The alignment tool takes, as its input, files in the FASTT format, a slight adaptation of the FASTA formatThe FASTA format is used to represent biological sequences, which consist of a very restricted alphabet and no spaces. In FASTT, we allow spaces to separate words. Software designed for FASTA sequences will generally not tolerate spaces; MAFFT, for example, requires you to replace spaces with another character in non-biological sequences. which is commonly used in bioinformatics. It looks like this:
>D
|| ātmā vastu svabhāvaś ca śarīraṃ tattvam ity api |
dravyam ity asya paryāyās tac ca nityam iti smṛtaṃ || ||
>K
|| ātmā vastu svabhāvaḥś ca śarīraṃ tatvam ity api |
dravyam ity asyāpardyāyās tac ca nityam iti smṛtaṃ ||
>V
ātmā vastu svabhāvaḥś ca śarīraṃ tattvaṃm ity api
dravyam ity asyāpardyāyās tac ca nityam iti smṛtaṃ
In this example, we have three versions of the same text (the first verse of the Dravyasamudeśa). Each text begins with the greater-than sign, followed by a siglum (D, K, or V here), a newline, and then the text.
Converting TEI XML to FASTT format
To produce files in this format from your XML transcriptions, you can use the converter. For example files, see the TEI XML transcriptions of the Dravyasamudddeśa.
Getting FASTT files from saktumiva.org
If we have transcribed our texts in TEI XML and uploaded them to saktumiva.org, then we can generate this file automatically, following these steps:
- Navigate to any one of the witnesses you want to include in your alignment, for example, Delhi University Library MS 5954.29.
- In the left-hand sidebar, under
Other witnesses
, select all of the other witnesses that you want in your alignment. You probably want to omit edited texts. - Click
Generate apparatus
. -
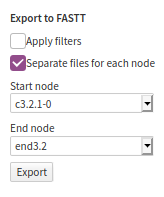
The FASTT export dialog. In the toolbar on the right-hand side, clickExport As...
and chooseFASTT
. This is an ad-hoc acronym forFASTA for Texts
. - In the options, select the range of the text that you want to align. The start node is the first paragraph/verse included, and the end node ls the last paragraph/verse.
- If the text is long, you will want to have separate files for each node (as in the tutorial files).
- You do not need to apply the filters, since the alignment tool will do that.
- Click
Export
.
You should end up with a zip archive full of .fas files, as in the directory example/fastt/. Files starting with c are paragraphs from the commentary, and files starting with v are verses from the Dravyasamuddeśa.
Preparing the alignment tool
There has been a lot of research on sequence alignment, and a lot of software has been developed to perform it, but, unfortunately, they are almost all exclusively focused on biological sequences.Apart from helayo mentioned here, other possibilities for aligning texts include CollateX or MAFFT with the --text option. In this repository, I have included a command-line tool to perform sequence alignment on Sanskrit texts, named helayo. It uses the Center Star algorithmAs implemented in the Data.Align.Affine package, also by the author. It has been forked from Data.Align, which follows Chin et al. 2003. Data.Align.Affine employs an affine gap penalty function. See Gusfield 1997, 243-244 or Sung 2009, 42-45 for a presentation of the affine gap penalty model. and is written in Haskell.
There are pre-built binaries for MacOS, Ubuntu Linux, and Windows. Download the appropriate helayo file from the dist directory of the GitHub repository.
Otherwise, you will need the Haskell Platform, which should include the stack build tool. Once you have installed that, go into the helayo/ directory and issue the following command:
stack build
Now, we are ready to align our texts.
Using the alignment tool
Helayo takes a FASTT file, removes spaces, punctuation, and sandhi, and aligns the texts in it. It produces a TEI XML file. In your terminal, navigate to the helayo/ directory. The basic syntax, using the substitution matrix (see below), is:If you have built helayo from source, replace ./helayo with stack exec helayo --.
./helayo -x substitution_matrix.csv [input file] > [output file]
For example, to align the first verse of the Dravyasamuddeśa, run
./helayo -x substitution_matrix.csv ../example/fastt/v3.2.1.fas > v3.2.1.xml
This will produce a file, called v3.2.1.xml, containing the aligned texts.
Alignment options: lemma size
Helayo can align texts as characters, akṣaras, or as words, using the -l character|aksara|word option. For example, to re-do the alignment of the first verse of the Dravyasamuddeśa as akṣaras, run
./helayo -x substitution_matrix.csv -l aksara ../example/fastt/v3.2.1.fas > v3.2.1-aksaras.xml
When experimenting with character, akṣara, or word lemmas, think about how your text might have been transmitted, and the mechanism by which changes occur. For example, if a scribe misinterprets a pṛṣṭhamātrā, then change is occuring on the character level. If a scribe writes rā instead of śa, then change is occuring on the akṣara level. If a scribe substitutes satyam with vidyām, then change is occuring on the word level.
Here is the same text aligned as characters, akṣaras, or words:
v a s t u s v a r ū p - - - - - a m a r th a k r i y ā k ā r i d r a v y a ṃ
v a s t a s v a l a k ṣ a - - ṇ a - - - - - k r i y ā k ā r i d r a v y a ṃ
v a s t u s v a l a k ṣ a m ā n a m a r th a k r i y ā k ā r i d r a v y a ṃ
va stu sva rū pa - - ma rtha kri yā kā ri dra vyaṃ
va sta sva la kṣa - ṇa - - kri yā kā ri dra vyaṃ
va stu sva la kṣa mā na ma rtha kri yā kā ri dra vyaṃ
vastu svarūpam arthakriyākāri dravyaṃ
vasta svalakṣaṇakriyākāri - dravyaṃ
vastu svalakṣamānam arthakriyākāri dravyaṃ
Alignment options: match, mismatch, and gap scoring
By default, the alignment is scored like this:Cartwright 2006, suggests an affine gap penalty function of GA(k) = 4 + k/4, but, in my work on the Prakīrṇaprakāśa manuscripts, this produced mismatches in places where gaps would have been more appropriate. The match score and the gap opening score have been adjusted accordingly.
match (-M) | 1 |
|---|---|
mismatch (-m) | -1 |
gap opening (-G) | -3 |
gap extension (-g) | -0.25 |
That is, a match between two readings, such as k and k (on a character level) or ka and ka (on an akṣara level) are given the score 1. A mismatch, such as k and p, is given the score -1. When working in aksara or word mode, mismatched readings have their total score divided by the length of the alignment; for example, ka and pa would be scored 0 (0/2), but ka and pi would be scored -2/2.
These options can be changed with the corresponding command-line option. For example, helayo -M 0.5 -m -2 would set the match score to 0.5 and the mismatch score to -2.
The substitution matrix
 The substitution matrix, colour-coded, displayed in LibreOffice Calc.
Helayo can optionally use a substitution matrix in order to improve the alignment. This is highly recommended. The matrix is a spreadsheet that gives different weights to different alignments. I have provided a simple matrix (
The substitution matrix, colour-coded, displayed in LibreOffice Calc.
Helayo can optionally use a substitution matrix in order to improve the alignment. This is highly recommended. The matrix is a spreadsheet that gives different weights to different alignments. I have provided a simple matrix (substitution_matrix.csv) that tries to align vowels with vowels and consonants with consonants:
| vowel/vowel mismatch | -1 |
|---|---|
| consonant/consonant mismatch | -1 |
| consonant/vowel mismatch | -1.25 |
For example, if the software tries to align t with g, that alignment will be scored as a mismatch, and given the penalty -1. But if it tries to align t with ā, that alignment will be scored even lower, at -1.25. Both are considered as mismatches, but mismatches between consonants and vowels are given a greater penalty.
You can edit the substition matrix in any spreadsheet software (LibreOffice Calc, Microsoft Excel, etc.) and save it as a csv file. See the csv2mafft/ directory for more details.
Working with multiple files
If you used the Separate files for each node option when exporting from saktumiva.org, then you might have a lot of files. You can run a simple command to align all the .fas files in a directory, and output them into a new directory. For example, to align all of the files in the example/fastt/ directory, using akṣara alignment, and output them to the example/xml/ directory, run
for f in ../example/fastt/*.fas; do ./helayo -x substitution_matrix.csv -l aksara ${f} > ../example/xml/$(basename $f fas)xml; done
WARNING: Especially when you have a lot of files, this is very slow. For the purposes of this tutorial, the example/xml/ directory has already been pre-filled with files aligned with the -l aksara option.
Using the matrix-editor
Now that the texts are aligned, you can check them in the matrix-editor. Simply use the version hosted on GitHub or, in the matrix-editor/ directory, start a simple web server. Load one of the XML files, and it will be displayed with each text as a row and each lemma as a column.
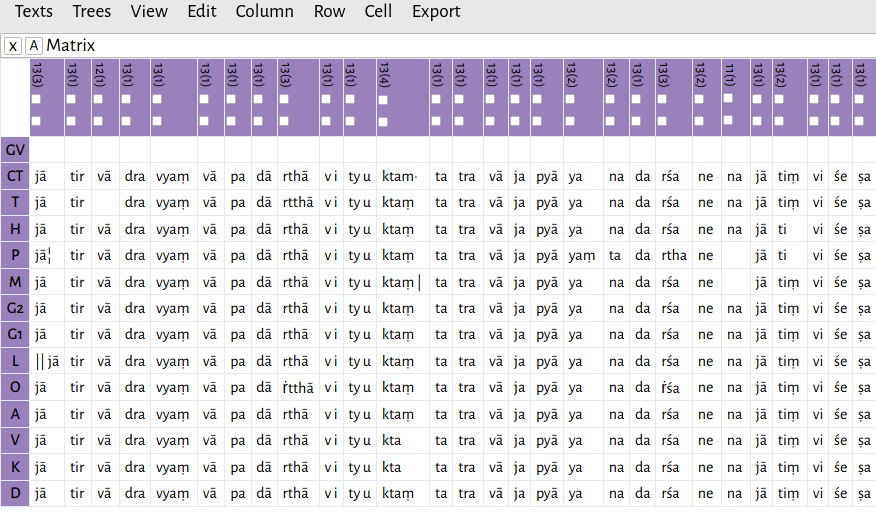
To see how the alignment tool has normalized the text, use the View menu and click Normalized.
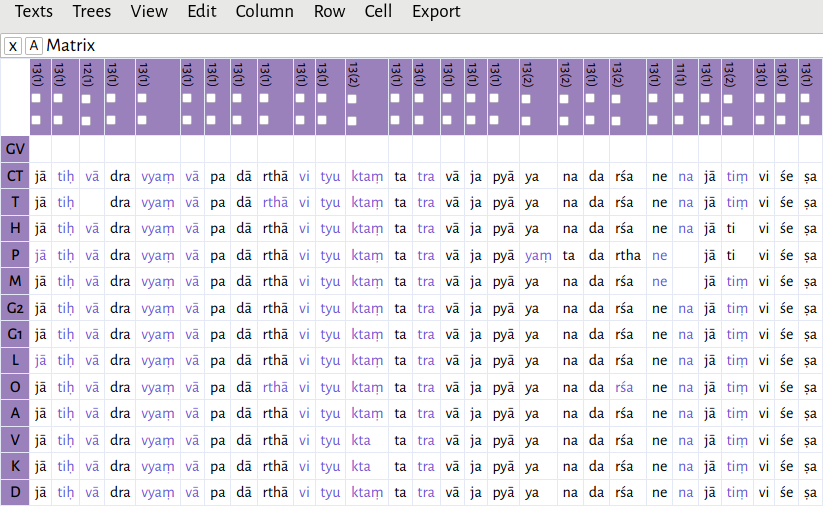
There may be some minor mis-alignments; you can edit an individual cell by clicking it and pressing Enter or using the menu option Cell -> Edit cell. In practice, the alignments are generally good enough as-is in order to produce a viable tree. Especially when the text is long, I have found the small mis-alignments have no effect. The Dravyasamuddeśa with commentary, split up as akṣaras, amounts to 7486 lemmata.
It is possible to export the alignment as a CSV file and edit it in any spreadsheet software, but, in doing so, you will lose the normalization data. You can, however, re-normalize the texts at any time by selecting columns and using the menu option Column -> (Re)normalize columns.
If you have done a character- or akṣara-based alignment, you can also ask the editor to try to group the columns into words, using the whitespace between characters as cues. This can be done by using the Group all words command in the Edit menu.
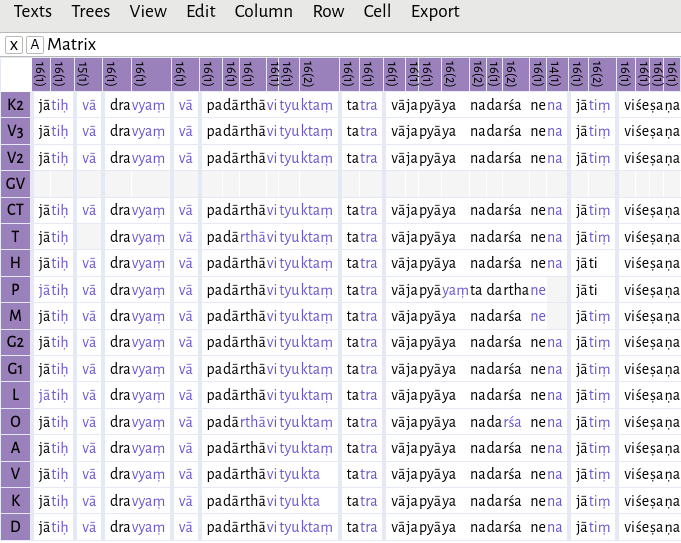
The results will differ depending on whether characters or akṣaras are being grouped. In this example,
pa dā rthā vi tyu ktaṃ
has been grouped together as padārthāvityuktaṃ
, even though, in the unnormalized text, there are spaces between padārthāv
, ity
, and uktaṃ
. The sequence vi is a single akṣara and cannot be split; the same applies to tyu. However, if we had done a character-based alignment,
p a d ā r th ā v i t y u k t a ṃ
the sequence could then be grouped as padārthāv
ity
uktaṃ
.
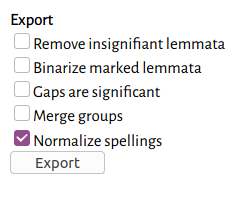
The NEXUS export dialog.
When you are satisfied with the alignment, you can export it as a NEXUS file, which can be used in phylogenetics software. If you have multiple files, you should open all of them together in the matrix-editor (use Ctrl-Click or Shift-Click to select multiple). Then use the Export menu and click NEXUS. You will probably want to use the normalized text in your phylogenetic analysis, so check the Normalize spellings box.
At this point, if you have done a character- or akṣara-based alignment, and you have used the Group all words function, you can also use the Merge groups option in order to try analyzing the alignment as words rather than characters or akṣaras.
The resulting NEXUS file is available in the GitHub repository at example/full-alignment.nex.
Building a tree
Now that we have a full alignment, we can use phylogenetics software to build a tree. There are many options for this, but in this tutorial we will use SplitsTree5.Other possibilities include PAUP*, PHYLIP, and Mesquite.
When you open the NEXUS file in SplitsTree, you will see a splits network representing the relationships between the witnesses.
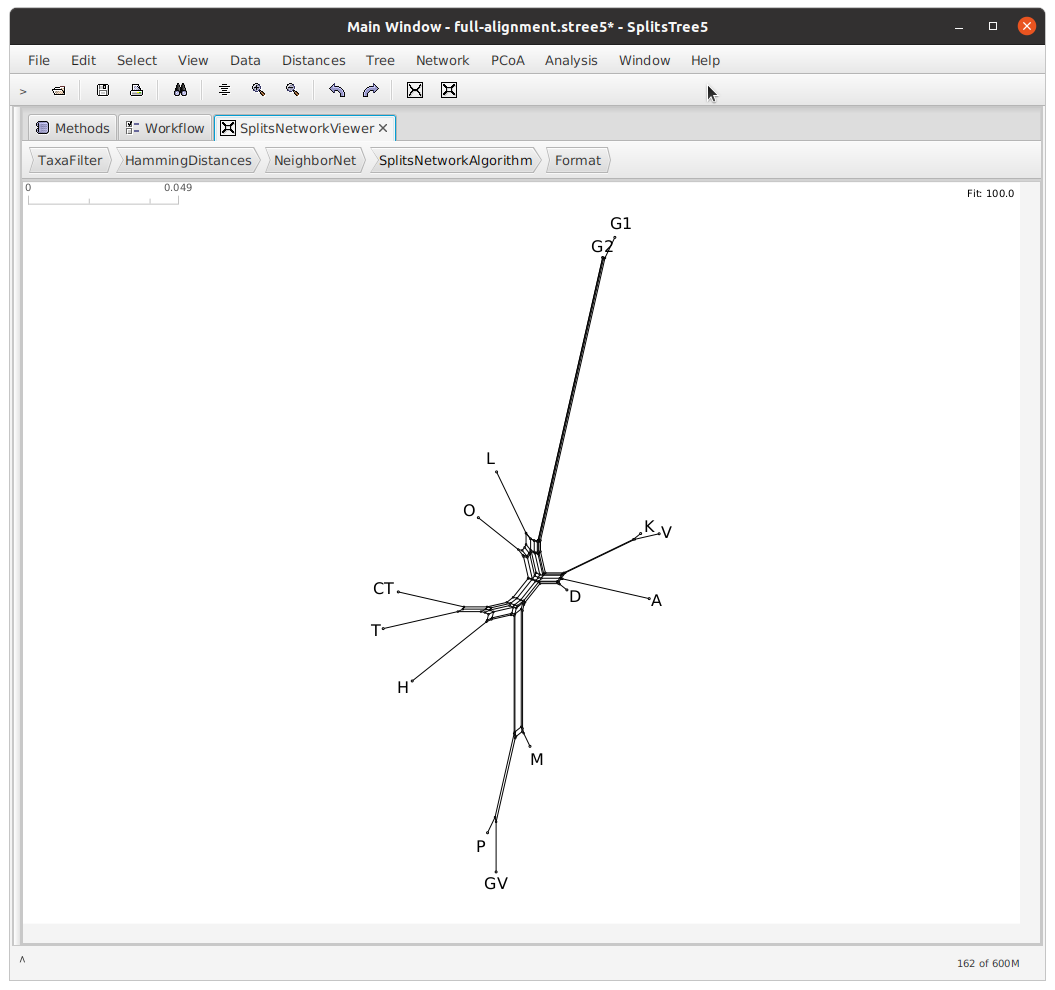
For more information on what this network represents, and how to interpret it, see Phillips-Rodriguez 2007 and Huson & Bryant 2006.
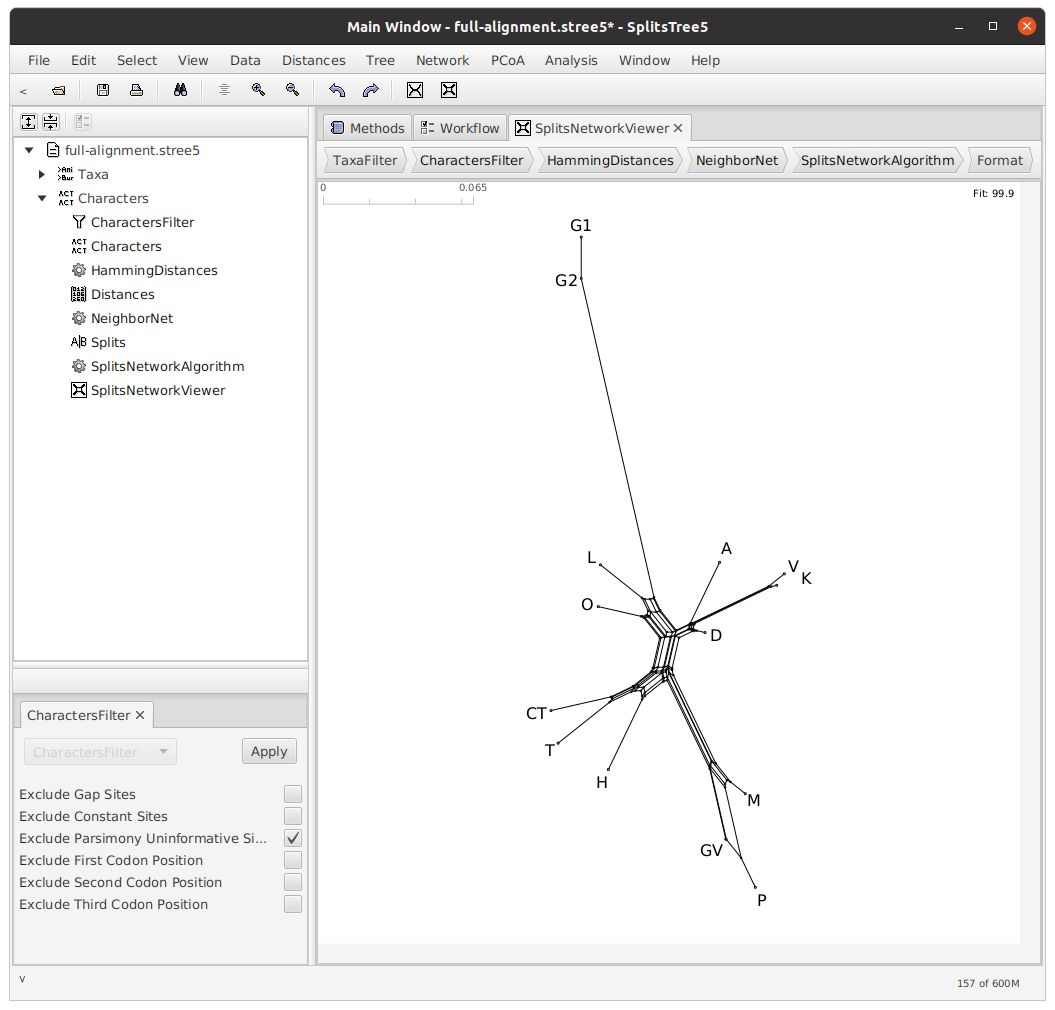
We can filter our lemmata so that only parsimony-informative lemmata are used in the construction of our network. Use the Data menu, click Filter Characters, and check the box next to Exclude Parsimony Uninformative Sites. Of the 7486 total lemmata, only 5499 were used in the creation of this network.
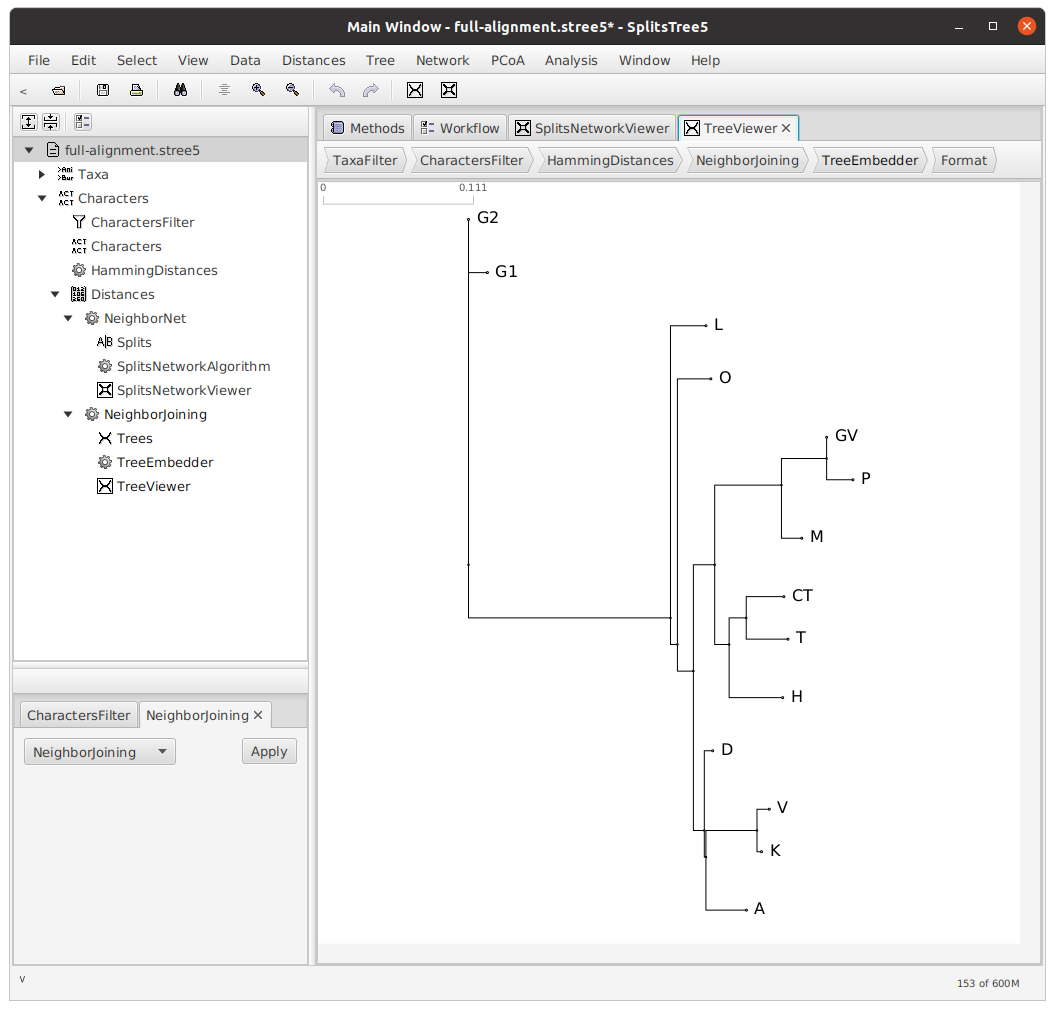
Now, we will use the Neighbor-joining algorithm to construct a tree from this network. Use the Tree menu and click NJ.
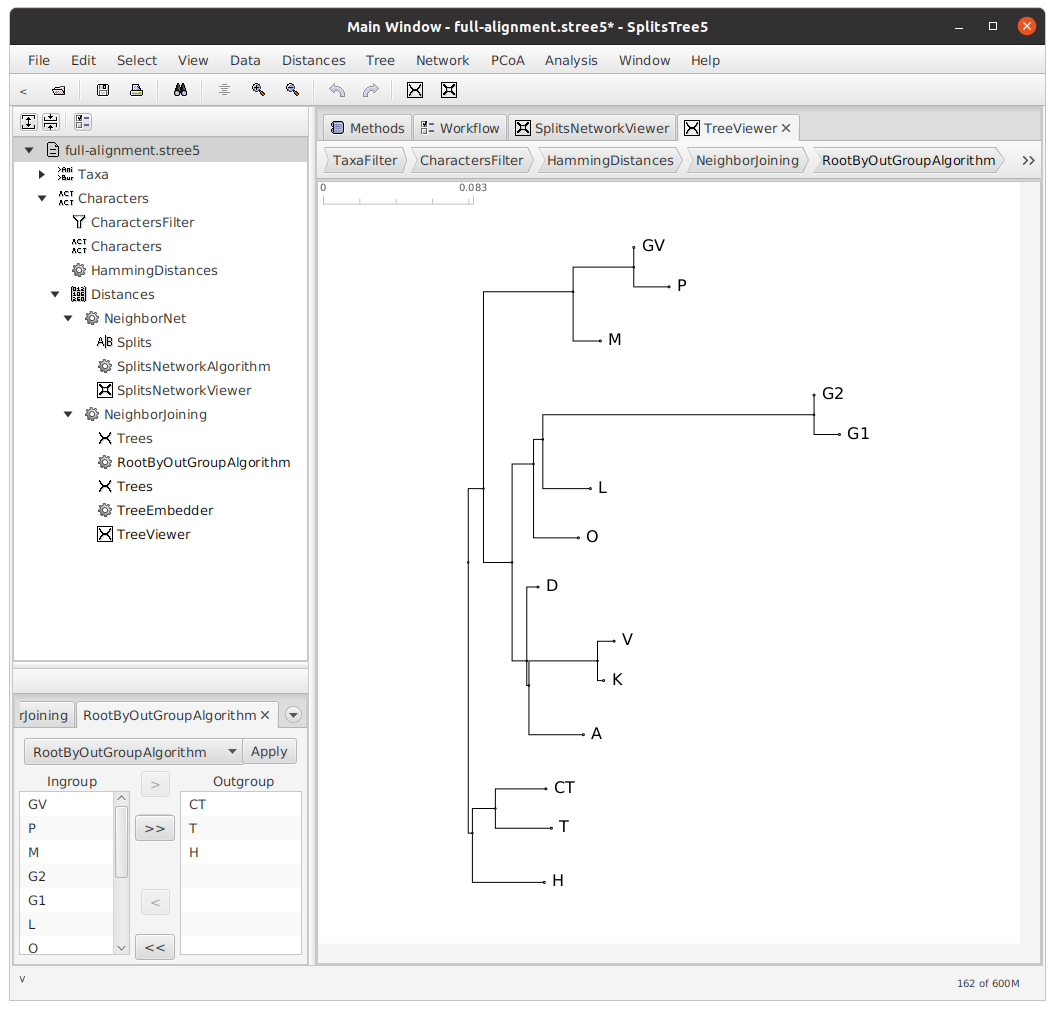
Rooting the tree
Next, we will root our tree.Rooting is not strictly necessary; for a discussion of unrooted trees, see Phillips-Rodriguez 2012. The decision about how to root a tree is dependent on many factors, and relies entirely on the expertise of the philolgist. In the case of this particular text tradition, I decided that there were two main branches — a southern branch, consisting of the witnesses CT, T, and H, and a northern branch, consisting of the remaining witnesses. Therefore, I rooted my tree with CT, T, and H as my outgroup.
Bootstrapping
In order to test how well our tree fits our data, we can do a simple test called bootstrapping. As Sandra L. Baldauf puts it,
This is done by taking random subsamples of the dataset, building trees from each of these and calculating the frequency with which the various parts of your tree are reproduced in each of these random subsamples. If group X is found in every subsample tree, then its bootstrap support is 100%, if its(!) found in only two-thirds of the subsample trees, its bootstrap support is 67%.
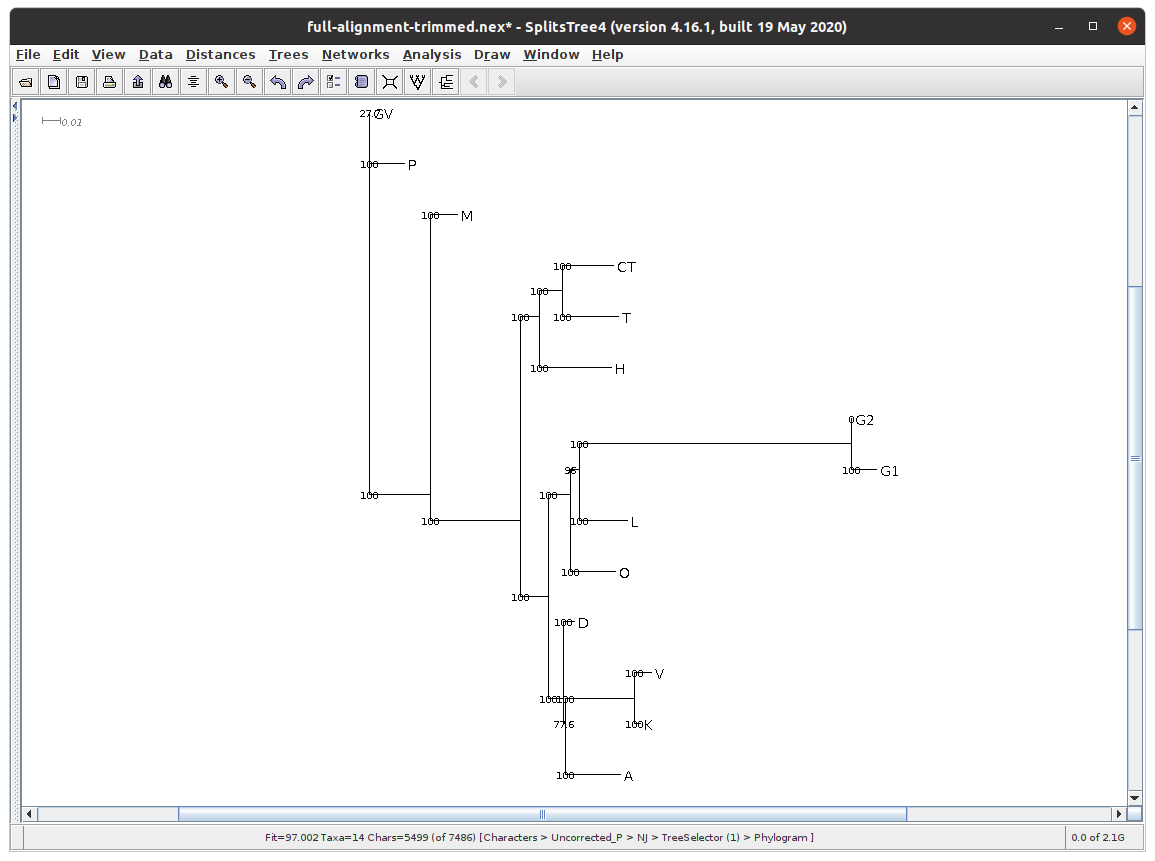
I used an older version of SplitsTree to perform bootstrapping (Analysis -> Bootstrap), since SplitsTree5 does not yet support it. As you can see, almost every branch has 100% or >90% bootstrap support. There are two exceptions:
- the branching of GV and P has only 27.7% support. This is very low, but GV is an extremely fragmentary manuscript, containing only the commentary on two verses, so this low score is understandable;
- the branching of D from the group A, K, and V has only 77.6% support. This is probably because D is a contaminated manuscript; the scribe/scholar has taken some readings from another branch and used them to correct his manuscript. In order to get better results, it may be better to omit D from our tree, or, alternatively, to use the ante correctionem readings from it, wherever legible.
Exporting to NeXML
In order to use our tree in the matrix-editor, we need to export it to NeXML format. Right-click on Trees in the sidebar (the second one, which is the rooted tree) and click Export....
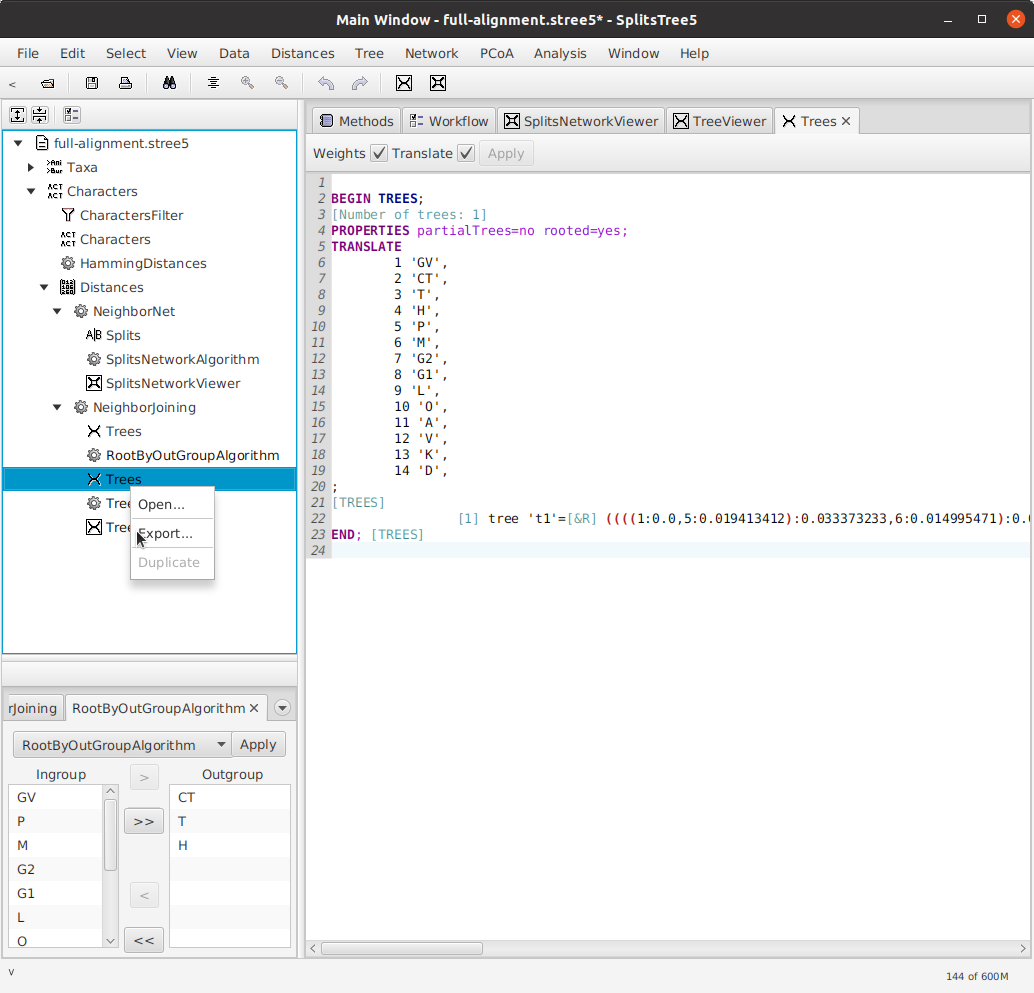
The exported tree from the example data can be found at example/trees/rooted-tree.xml.Earlier versions of SplitsTree5 were missing the top-level <nex:nexml> element, but this has been fixed since version 5_2_22-beta.
Reconstructing a text
Reconstructing a single lemma
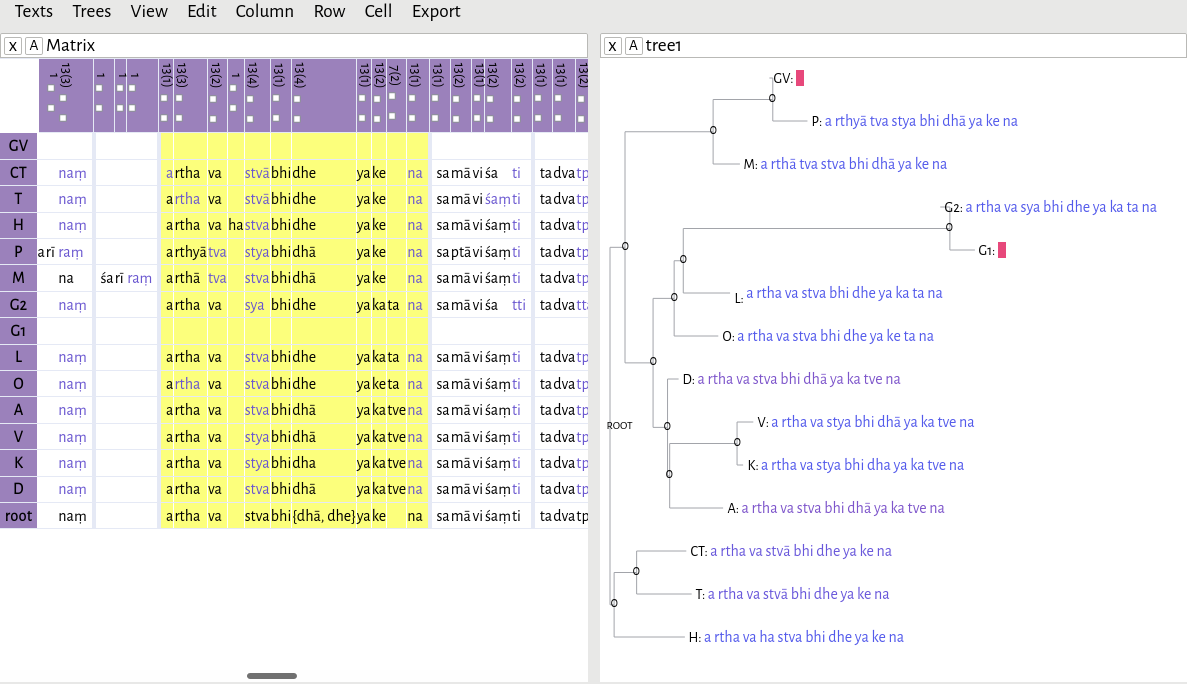
Now that we have prepared the tree, we can open it in the matrix-editor to do some reconstruction. First, in the matrix-editor, open up one or more of the aligned files again. It is probably easier to do them one at a time, or a couple at a time. Then, from the Trees menu, open the NeXML tree.
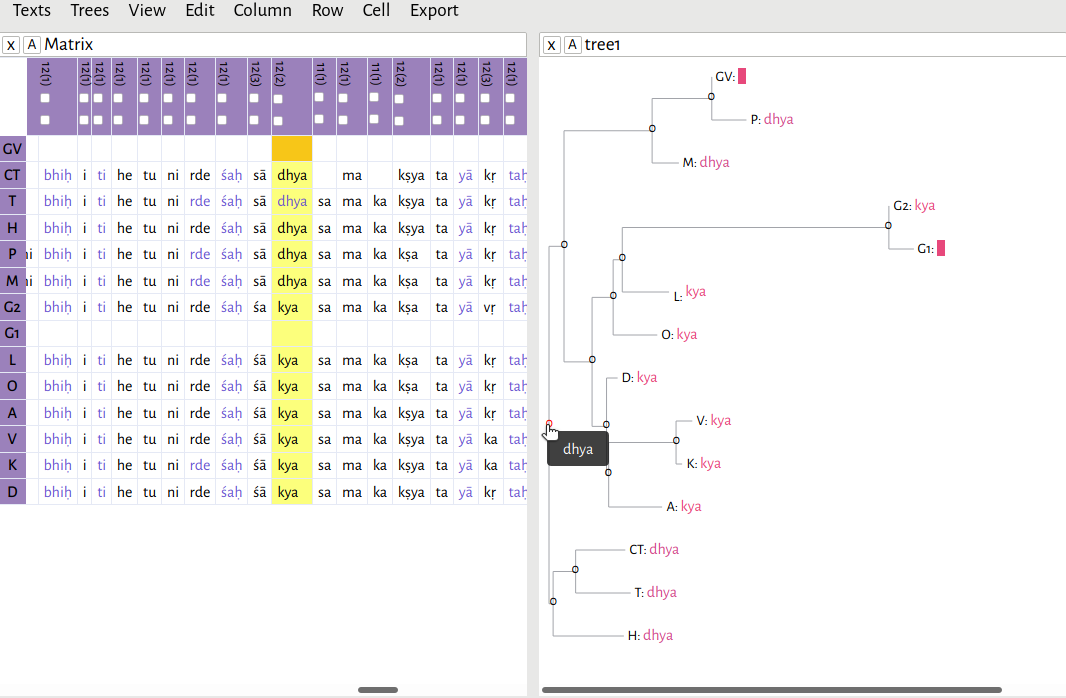
In this example, I have opened the file c3.2.4-2.xml, and then opened rooted-tree.xml. As you can see, if you hover your mouse over any one of the nodes in the tree, the editor will give you a reconstructed reading based on Fitch's algorithm.
As I mentioned previously, I hypothesized there to be two main branches in this text tradition: a southern branch, consisting of witnesses CT, T, and H, and a northern branch. Here, the southern branch consistently reads dhya
. The northern branch is split between dhya
and kya
.For a discussion of this lemma and its significance, see Li 2018, 48n118. Because of the way I've rooted the tree, the algorithm has decided that dhya
is the correct reading, since it has support in both main branches. But what if I had decided that all of the witnesses with the reading dhya
form one outgroup?
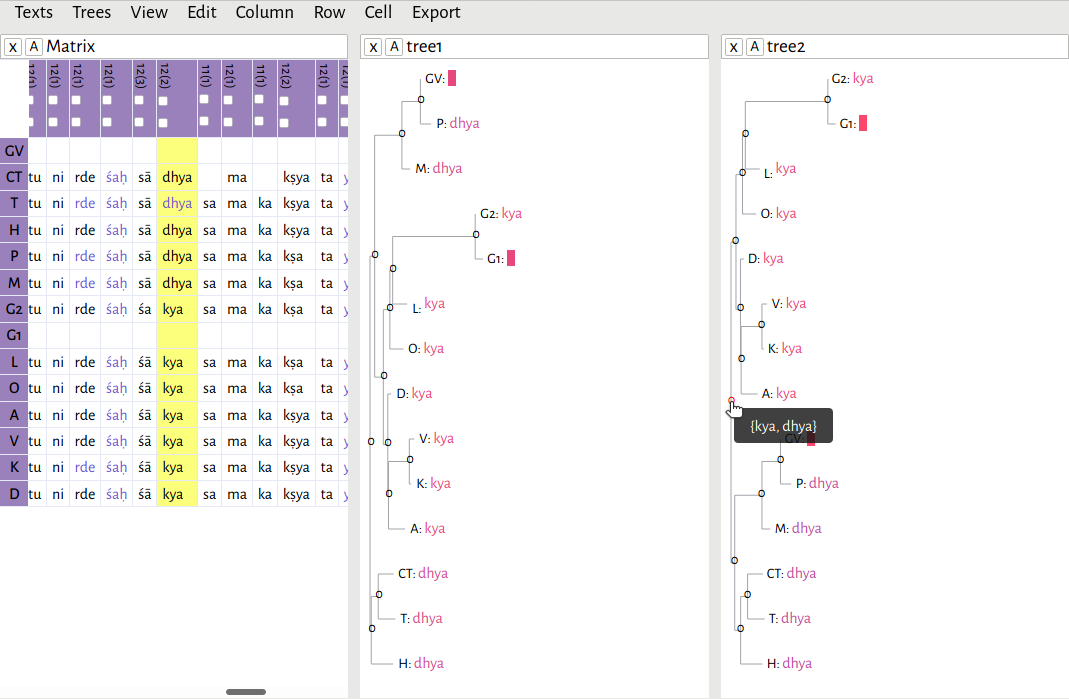
I re-rooted the tree, this time using CT, T, H, M, P, and GV as my outgroup (this file is available at example/trees/alternative-rooted-tree.xml). Now, both readings, kya
and dhya
, are equally weighted.This is exactly the situation described in Maas 1958, 6: The reconstruction of α is a different matter. If its tradition has two branches only, β and γ, and β and γ agree, we have the text of α. If they do not agree, then either of the two readings may be the text of α; we have here variants, between which it is not possible to decide on the lines of our procedure hitherto.
Reconstructing an entire passage
Instead of reconstructing the text lemma-by-lemma, we can ask the software to reconstruct the entire text for us. First make sure you are using the normalized readings (View -> Normalized). Then, simply click one node. Presumably, we will want to reconstruct the root — that is, what we hypothesize to be the oldest reachable state of the text, based solely on the manuscript evidence. However, you can also reconstruct the text at any later stage of its evolution; for example, if you wish to reconstruct the northern recension of the text, simply click on the node that groups all the northern witnesses together. Here, for the sake of an example, I have chosen the root.
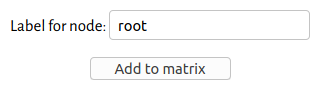
The node reconstruction dialog box.
Give the node a label; here I have labelled it root
. Then click Add to matrix. This will add a new row to the bottom of the matrix, with the reconstructed text. It may take up to a few minutes to complete.
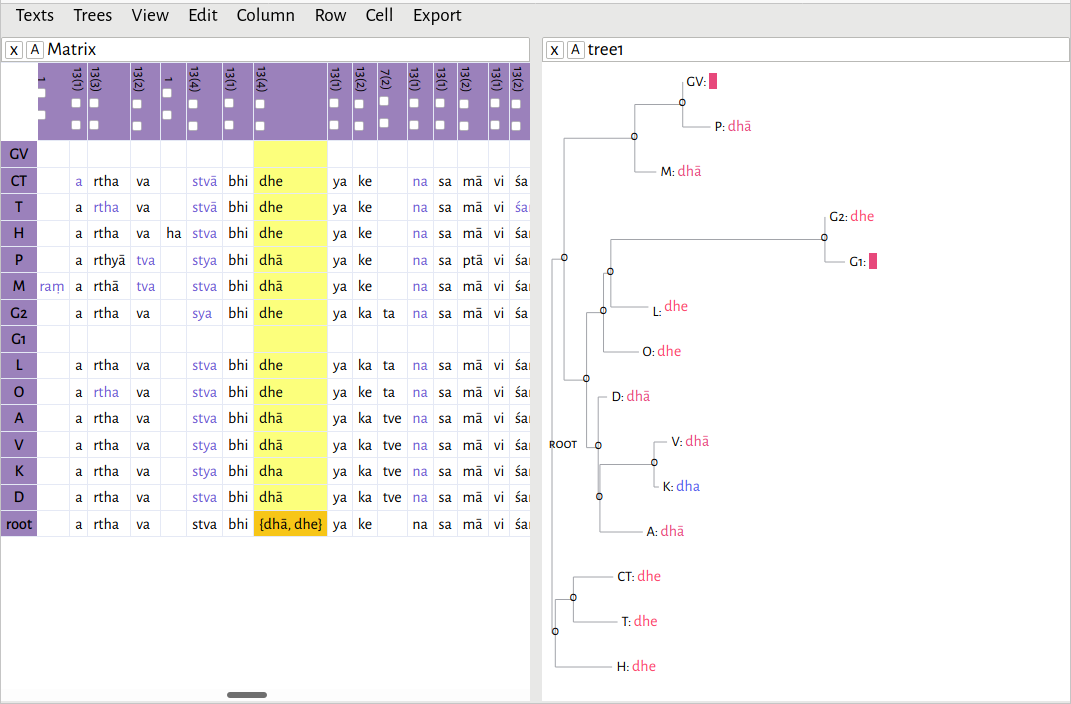
Where the algorithm is able to come up with a definitive reading, it will be displayed in the cell. But if there is more than one possibility, it will give you all possible readings, like so: {dhā, dhe}
. In this case, there are two, equally-weighted possibilities: dhā
and dhe
. Thus, the word here could equally be abhidhāyakena
or abhidheyakena
. Now, it is up to the philologist to decide the correct reading. But note that, up until this point, every step in the process has been reproducible. In a case such as this, where the reading cannot be determined stematically, it would be appropriate for the editor to include a note in their critical text, stating their choice, and, if, the reasoning behind it is not immediately obvious, a justification.
Exporting a critical apparatus
After reconstructing a passage, the text can be exported in TEI XML format, as a text with variant readings. If you have done the alignment as characters or akṣaras, you will probably want to group them into larger lemmata before exporting. You can do this by using the Group all words command in the Edit menu, if you haven't already.

The TEI apparatus export dialog.
If you want to change the groups, you can select columns and use the Column -> Ungroup columns and Column -> Group columns commands to reform them. Once you are satisfied, you can export the text in TEI XML format, by choosing TEI apparatus from the Export menu. There, you will be able to choose a base text (probably the reconstructed text). You will want to make sure that Merge groups is checked, so that the apparatus of variants is constructed based on the grouped "words" rather than on each individual character or akṣara.
The resulting TEI XML file can then be edited — for example, you may wish to prune the apparatus so that only the most important variants are shown. Since all of the manuscript witnesses have been transcribed already and can be compared using tools like saktumiva.org, this curated
apparatus can be a useful way to focus on certain textual issues, while still having a fully positive apparatus available to the reader.
Going further
Both the software described in this document, as well as this document itself, are in development, and there is much more work to be done. The formal algorithms that we are using date from the 70's, and the techniques — considering Lachmann — from the 19th century. But the more data that we produce, in the form of diplomatic transcriptions of manuscripts and stemmatic studies of text traditions, the more we can start to create models of how texts evolve over time, and the better equipped we will be to devise better algorithms to explore them. In this document, I have mainly described the technical elements of textual reconstruction; I have only touched briefly on the philological considerations, and given mere hints of the kinds of research questions that we can address using these techniques.
Bibliography
Baldauf, Sandra L. 2003. Phylogeny for the faint of heart: a tutorial.
Trends in Genetics 19(6): 345-351. https://www.researchgate.net/publication/10712132_Phylogeny_for_the_faint_of_heart_A_tutorial.
Cartwright, Reed A. 2006. Logarithmic gap costs decrease alignment accuracy.
BMC bioinformatics 7(527). https://doi.org/10.1186/1471-2105-7-527.
Chin, Francis Y. L. et al. 2003. Efficient Constrained Multiple Sequence Alignment with Performance Guarantee.
In Computational Systems Bioinformatics, Proceedings of the 2003 IEEE Bioinformatics Conferece. Stanford, CA: IEEE. https://ieeexplore.ieee.org/document/1227334.
Fitch, Walter M. 1971. Defining the course of Evolution: Minimum change for a specific tree topology.
Systematic Zoology 20:406–416. https://www.jstor.org/stable/2412116.
Gusfield, Dan. 1997. Algorithms on Strings, Trees, and Sequences. Cambridge: Cambridge University Press. https://doi.org/10.1017/CBO9780511574931.
Huson, Daniel H. & Bryant, David. 2006. Application of Phylogenetic Networks in Evolutionary Studies.
Molecular Biology and Evolution 23(2): 254–267. https://academic.oup.com/mbe/article/23/2/254/1118872.
Katoh, Kazutaka, Kazuharu Misawa, Kei-ichi Kuma, & Takashi Miyata. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform.
Nucleic Acids Research 30(14): 3059–3066. https://doi.org/10.1093/nar/gkf436.
Li, Charles. 2017. Critical diplomatic editing: Applying text-critical principles as algorithms.
In Advances in Digital Scholarly Editing, edited by P. Boot et al, 305-310. Leiden: Sidestone Press. https://hcommons.org/deposits/item/hc:18259/.
Li, Charles. 2018. Limits of the real: A hypertext critical edition and translation of Bhartṛhari’s Dravyasamuddeśa, with the commentary of Helārāja.
PhD thesis, University of Cambridge. https://www.repository.cam.ac.uk/handle/1810/284085.
Maas, Paul. 1958. Textual Criticism. Translated by B. Flower. Oxford: Clarendon Press.
Phillips-Rodriguez, Wendy J. 2007. Electronic Techniques of Textual Analysis and Edition for Ancient Texts: an Exploration of the Phylogeny of the Dyūtaparvan.
PhD thesis, University of Cambridge.
Phillips-Rodriguez, Wendy J. 2012. Unrooted trees: A way around the dilemma of recension.
In Battle, Bards, Brahmins (Papers of the 13th World Sanskrit Conference) edited by J. Brockington, 217-230. Delhi: Motilal Banarsidass.
Sung Wing-Kin. 2009. Algorithms in Bioinformatics: A Practical Introduction. London: CRC Press.
Trovato, Paolo. 2014. Everything You Always Wanted to Know about Lachmann’s Method. Translated by F. Poole. Padova: liberiauniversitaria.it.